Archives
- 2019-01
- 2019-04
- 2019-05
- 2019-06
- 2019-07
- 2019-08
- 2019-09
- 2019-10
- 2019-11
- 2019-12
- 2020-01
- 2020-02
- 2020-03
- 2020-04
- 2020-05
- 2020-06
- 2020-07
- 2020-08
- 2020-09
- 2020-10
- 2020-11
- 2020-12
- 2021-01
- 2021-02
- 2021-03
- 2021-04
- 2021-05
- 2021-06
- 2021-07
- 2021-08
- 2021-09
- 2021-10
- 2021-11
- 2021-12
- 2022-01
- 2022-02
- 2022-03
- 2022-04
- 2022-05
- 2022-06
- 2022-07
- 2022-08
- 2022-09
- 2022-10
- 2022-11
- 2022-12
- 2023-01
- 2023-02
- 2023-03
- 2023-04
- 2023-05
- 2023-06
- 2023-08
- 2023-09
- 2023-10
- 2023-11
- 2023-12
- 2024-01
- 2024-02
- 2024-03
- 2024-04
- 2024-05
- 2024-06
- 2024-07
- 2024-08
- 2024-09
- 2024-10
- 2024-11
- 2024-12
- 2025-01
- 2025-02
- 2025-03
-
Scutellaria moniliorrhiza Komarov Labiatae is a perennial he
2025-03-03
Scutellaria moniliorrhiza Komarov (Labiatae) is a perennial herb and mainly distributed in Jilin province, China. In traditional Chinese medicine, the whole plants have been used to clear away heat-evil, expel superficial evils, eliminate stasis and reduce edema (He et al., 2012). In previous phytoc
-
The finding that OCT is expressed in the nucleus
2025-03-03
The finding that OCT3 is expressed in the nucleus accumbens (Gasser et al., 2009) suggests that inhibition of this transporter may underlie previously described effects of glucocorticoids on extracellular DA concentrations and indicates that DA clearance in this region is not mediated exclusively by
-
Adiponectin also works on AdipoR inducing
2025-03-03
Adiponectin also works on AdipoR1, inducing extracellular Ca2+ influx necessary for activation of Ca2+/calmodulin-dependent protein kinase kinase (CaMKK)β, AMPK (Fig. 1). This step is then followed by activation of SirT1 and increased expression and decreased acetylation of PPARγ coactivator (PGC)-1
-
Reverse transcription enzyme We have previously shown that t
2025-03-03
We have previously shown that the antinociceptive effect of tramadol, an analgesic that, like paracetamol is able to increase serotonin levels within CNS, is potentiated or antagonized respectively by a 5-HT1A/B nonspecific Reverse transcription enzyme blockade or activation (Rojas-Corrales et al.,
-
br Effective therapeutic window of AR actions on ROP Retinal
2025-03-03
Effective therapeutic window of AR actions on ROP Retinal vasculature undergoes critical developmental changes postnatally: from P7 onward the superficial procyanidin start sprouting vertically in retina to form first the deep then the intermediated vascular plexus in the retina of C57BL/6 mice
-
To reconstruct the amino acid sequence of
2025-03-03
To reconstruct the amino Z-Ligustilide sequence of an ancestral β subunit a molecular phylogeny is required, which is inferred from a β subunit sequence alignment. To build this alignment we searched the NCBI database for proteins similar in sequence to the human muscle-type β subunit precursor pro
-
Two critical determinants of receptor trafficking are
2025-03-01
Two critical determinants of receptor trafficking are found within the GABAB1 cytoplasmic tail: the di-leucine internalization signal (EKSRLL) (Margeta-Mitrovic et al., 2000, Restituito et al., 2005) and the ER retention signal (RSRR) (Calver et al., 2001, Margeta-Mitrovic et al., 2000, Pagano et al
-
daunorubicin br Introducing toll like receptors TLRs are
2025-03-01
Introducing toll like receptors TLRs are the most important receptors which are expressed on the cytoplasmic and internal vesicles membrane. They have a similar structure including a leucine-rich repeats (LRRs) domain which is out of daunorubicin membrane, a hydrophobic transmembrane and a toll/
-
br HMG proteins mobile modulators of chromatin structure and
2025-03-01
HMG proteins: mobile modulators of chromatin structure and cellular phenotype The term “High Mobility Group” was originally coined for the HMG proteins because of their unusual solubility properties, their small size and their rapid mobility, relative to other chromatin proteins, during gel elect
-
There are increasing repeated reports
2025-03-01

There are increasing repeated reports of amphotericin B-resistance in pathogenic fungi including Candida spp. [35], [36]. Nolte et al. characterized some fluconazole and amphotericin B-resistant Candida albicans isolates from leukemia patients [37]. Fan-Havard et al. (1991) have shown multi-antifung
-
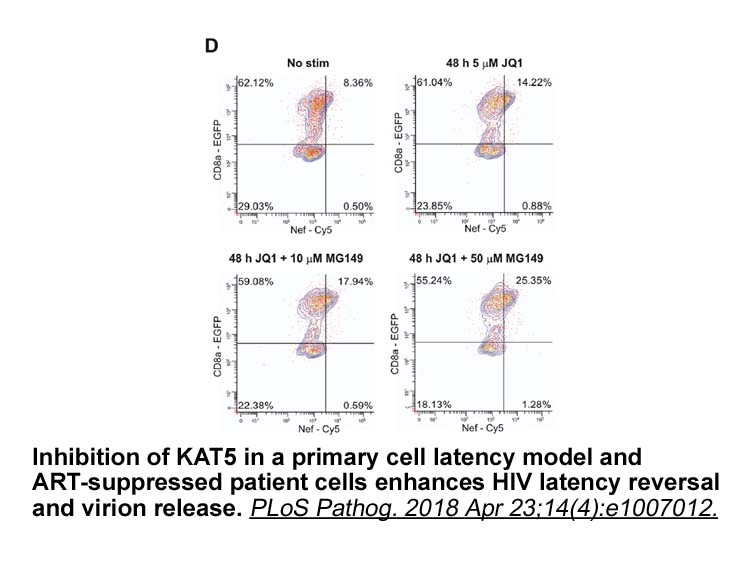
br Concluding Remarks and Future Perspectives Herein we
2025-03-01
Concluding Remarks and Future Perspectives Herein, we have highlighted our current understanding of the role of the LKB1-AMPK pathway and its related kinases in β cell biology. β cell-specific genetic models have been particularly useful in delineating precise roles for individual family members
-
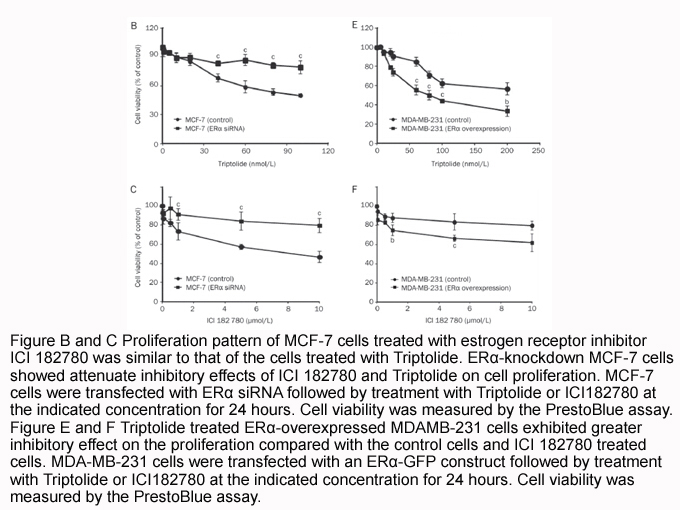
BTB06584 Overexpression of AR can lead to sorbitol accumulat
2025-03-01
Overexpression of AR can lead to sorbitol accumulation, leading to diabetic complications (Ransohoff and Cardona, 2010). Therefore, AR is an important enzyme in the growth and development of organisms. With the aim of inhibiting the activity of AR in diabetic patients, screening for inhibitors and i
-
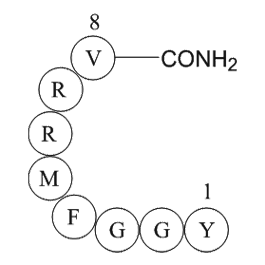
Tropomyosi http www apexbt com media diy images wb B
2025-03-01
Tropomyosin-related kinases (Trks) play crucial role in neurotrophin-mediated cellular activities, including neuronal differentiation, survival, and synaptic function [37] through activating downstream signaling mediators PI3K and Akt [38]. In a therapeutic context, Akt has been shown to mediate str
-
Growing evidence indicates that GRKs can exert different
2025-03-01
Growing evidence indicates that GRKs can exert different effects within the cell depending on the stimulus, cell type, and localization [97], [121]. In this sense, we were the first to demonstrate a mitochondrial localization for GRK2 [122], later confirmed by other investigators [123], establishing
-
While A Rs communicate mainly with the D
2025-03-01
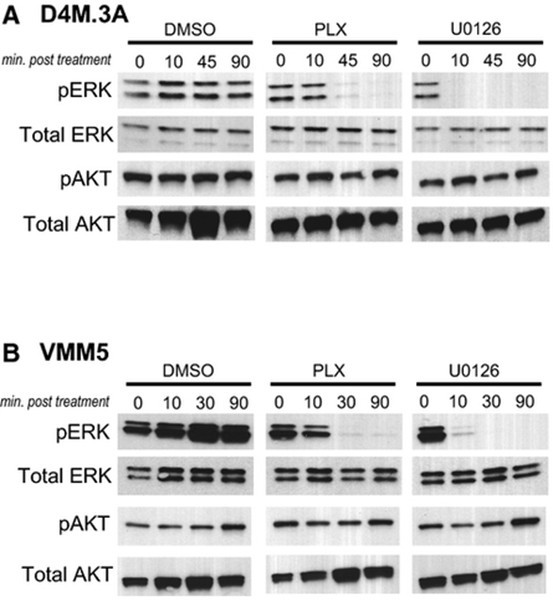
While A1Rs communicate mainly with the D1R subtype [36], A2AR and D2R interaction occurs mainly in basal ganglia. D2Rs colocalizes with A2ARs in this azacytidine area where they are preferentially localized postsynaptically in the soma and dendrites of GABAergic striatopallidal neurons [69]. This i